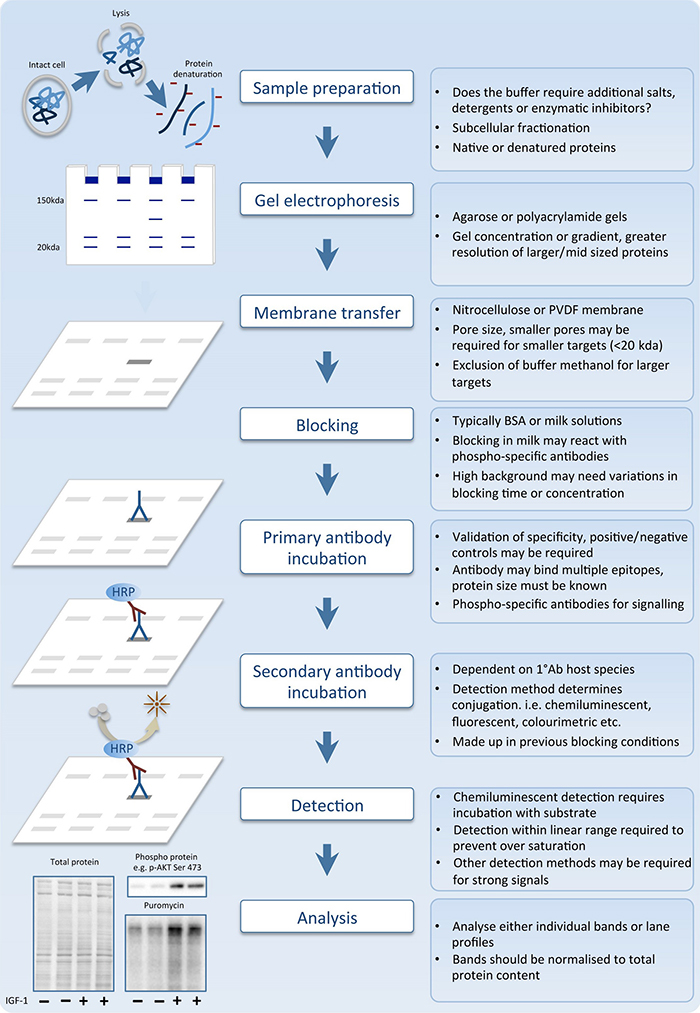
Fig. 1. The sequential stages of the western blot process.
Photo courtesy of”
https://onlinelibrary.wiley.com/doi/10.1111/sms.12702/full
The 8 basic steps of the science of Western blotting have remained the same for the last 40 years.
1. First, sample preparation: lysing and denaturing the proteins.
2. Next, gel electrophoresis.
3. Then membrane transfer.
4. After that blocking.
5. Then incubating the primary antibody.
6. And, then incubating the secondary antibody.
7. Next detection.
8. And, finally analysis.
But, while the steps haven’t changed, new methods of automation for the blocking, washing and incubating the primary and secondary antibodies have increased reliability and reproducibility in Western blot research.
Photo courtesy of:
https://www.ruf.rice.edu/~bioslabs/studies/sds-page/sdsgoofs.html
However even with automation, not every blot turns out the way we’d like. As a matter of fact, Rice University has an online “Hall of Shame” that presents Western blot goofs. The site discusses the joys and “oh boys” of some of the most colorful Western blot failures. Sometimes blot failures can be attributed to bad antibodies; however, this article relates that variations in western blot assay results occur because of errors in manual processing.
So, to assist you, whether you do manual or automated processing, we’re providing this:
Western Blot Troubleshooting Guide (click to download the PDF)
Problem
Diffuse Bands
Possible Cause
1. Antibody concentration too high
2. Excess protein on gel
3. Protein transfer was too fast and/or the gel was over-heated during electrophoresis
Solution
Problem
Nonspecific bands
Possible Cause
1. Non-specific binding to immobilized protein bands caused by SDS
2. Gradually accumulative differences of protein expression profiles due to frequent passage of cell lines
3. Degraded protein sample
4. Presence of new proteins or different splice variants that share similar epitopes
5. Presence of impurities with antibody
6. Formation of multi-mer on protein target
7. Formation of different protein subtypes which have different molecular weights
8. Presence of multi-modifier locus in protein
9. Primary antibody concentration too high
10. Non-specific bands caused by secondary antibody
11. Excess protein on gel
12. Insufficient washing
13. Blocking problem
Solution
Problem
Weak or no signal from the blot
Possible Cause
1. Detection step missed or detection reagents not working.
2. Insufficient incubation with detection reagent.
3. Poor or incomplete transfer
4. Protein of interest ran off the gel
5. Incorrect reagents added or incorrect containers are filled
6. Sample is too dilute.
7. Poor retention of proteins or the protein is weakly bound to membrane.
8. Inactive or overly dilute primary or secondary antibody.
Solution
Problem
High background on the blot
Possible Cause
1. Film overexposed or became wet during exposure
2. Short blocking time or washing intensity
3. High concentration of primary and/or secondary antibody
4. Protein is overloaded
5. Membrane, solutions, trays, or antibody containers are contaminated.
Solution
Problem
Non-specific binding too high
Possible Cause
1. Insufficient removal of SDS or weakly bound proteins from membrane after blotting.
2. Short blocking time
3. Affinity of the primary antibody for the protein standards
4. Protein is overloaded
Solution
If you need technical help with your BlotCycler™ device that’s not discussed here in our Western Blot Troubleshooting Guide, please contact us 1-888-490-4443. extension 1.


